gaussian 09w 64位(高斯09程序包)
vd.01 破解版_(含中文手册+安装教程)
- 软件大小:188.00 MB
- 软件语言:中文
- 软件类型:国外软件 / 教育学习
- 软件授权: 破解软件
- 更新时间:2017-07-18 16:30:32
- 软件等级:

- 软件厂商: -
- 应用平台:WinXP, Win7, Win8, Win10
- 软件官网: 暂无
相关软件

宝宝树孕育pc版v9.10.1 官方版
77.58 MB/中文/10.0

橙果果软件v3.2.1 安卓版
38.00 MB/中文/10.0

AceReader elite(教育辅助阅读软件)v10.0.4 特别版
48.21 MB/英文/10.0

医学下载课堂软件v1.0.0.54 免费电脑版
70.18 MB/中文/10.0

宏鹏教育软件v1.5.0 官网安卓版
18.12 MB/中文/10.0

软件介绍人气软件精品推荐相关文章网友评论下载地址
gaussian 09 win 64位(高斯09程序包)是一款量子化学相关且功能强大的综合软件包,相当于一个数据库类的庞大计算工具,应用领域挺广的,推荐给有需要的朋友下载使用!
gaussian 09 破解版简介
gaussian 09(高斯09)是目前计算化学领域内最流行、应用范围最广的综合性量子化学计算程序包。gaussian 09基于量子力学而开发,它致力于把量子力学理论应用于实际问题,它可以通过一些基本命令验证和预测目标体系几乎所有的性质。此外,可视化软件GaussView的发布及计算机的快速发展更是大大降低了理论计算的门槛,使得各领域研究者能够轻松使用Gaussian研究和分析各种科学问题。同时gaussian 09的电子结构的建模提供了先进的功能。高斯09是用于多种计算机系统。所有版本的高斯09包含每一个科学/建模功能,并没有施加任何人为的限制对其他比你的计算资源和耐心的计算。gaussian 09拥有着众多的实用功能,例如分子和反应综合调查,在这里用户可以彻底调查自己感兴趣的化学问题。例如,你不仅可以减少分子结构迅速可靠,还可以预测过渡态结构,并确认位于固定点实际上是极小值和过渡态。可以去计算反应路径的内在反应坐标(IRC)和确定反应物和产物的一个给定的过渡结构连接。一旦你有一个完整的势能面图,反应的能量和障碍可以准确地预测。同时gaussian 09还有着模拟核磁共振功能,gaussian 09继续加强计划的核磁共振功能。自旋-自旋耦合常数产生定量的最困难的光谱数据。的计算精度是高度依赖于基础设置。而量子化学的标准基组是价电子的发达,一个更复杂的电子密度接近核的描述是预测费米接触需要(FC)长期(通常的自旋-自旋耦合常数的最大组成部分)。更多功能,用户自行下载gaussian 09探索吧。
PS:小编说的windows系统是基于用户在安装linux虚拟机的情况下才可正常运行的哦,没有虚拟机的用户可以到此下载:http://www.itmop.com/downinfo/2685.html

特点
1、自动平衡计算
2、自动优化频率或单点能量
3、可以轻松添加、删除冻结,区分冗余内部坐标。
4、简化同位素替代路线部分和温度/压力规范
5、原子冻结的类型、片段、ONIOM层,PDB残留在优化
6、简化片段分子规范定义
7、更多的可重新起动的工作类型
8、原子在优化冻结类型、片段、ONIOM层和/或残渣
9、QST2 / QST3自动化过渡结构优化
10、选择和排序正常模式在感兴趣的频率计算,保存和阅读正常模式
11、从不同的检查点文件检索的信息比用于计算
gaussian09安装教程
1、(使用root登陆)拷贝高斯安装文件g03.tar.gz到/home/viger/gaussian03-64/目录下:连接你的U盘,用CP命令复制
2、用如下命令解压复制到linux的g03软件包:
tar -xvfz g03.tar.gz (tar为解压命令,-xvfz为选项)
解压后得到g03文件夹。
3、进行权限设置:
chown -R viger gaussian03-64
chgrp -R users gaussian03-64
chmod -R g+w gaussian03-64
这步是设置普通用户使用gaussian的权限,不做会造成普通用户不能运行gaussian等后果
4、用户路径下添加.cshrc文件:
vi /home/viger/.cshrc
按“i”变为插入模式
添加以下几行:
g03root /home/viger/gaussian03-64 (确认高斯路径)
source $g03root/g03/bsd/g03.login
保存修改:按Esc建回到一般模式(左下角插入标志消失),然后输入“:x”回车进行保存,就退出.cshrc了。具体参照vi命令的使用方法!
然后运行source /home/viger/.cshrc 如不提示错误就好了。
5、cd进入g03文件然后进入其bsd文件夹,运行./install
(若不能正常安装,可能是原文件被破坏,请重新copy原文件)
6、(viger登陆) 在/home/viger/Gaussian/g03/下建立scratch文件夹(这步可以不做)
进入/home/viger/Gaussian/g03/,mkdir scratch
7、可以建一个小分子的文件,如水的计算文件h20.com文件:
vi h2o.com
输入计算文件
保存
然后运行命令:g03 h20.com&
正常运行说明安装成功
gaussian 09w 64位(高斯09程序包)功能介绍
1、分子和反应综合调查
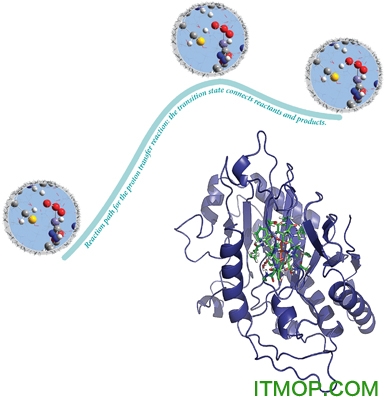
高斯09,你可以彻底调查你感兴趣的化学问题。例如,你不仅可以减少分子结构迅速可靠,还可以预测过渡态结构,并确认位于固定点实际上是极小值和过渡态。你可以去计算反应路径的内在反应坐标(IRC)和确定反应物和产物的一个给定的过渡结构连接。一旦你有一个完整的势能面图,反应的能量和障碍可以准确地预测。研究人员使用这些基本功能的高斯09研究异青霉素N合成酶(IPN),一个家庭的单核非血红素铁酶的成员(图在下一个图像的右下角)。过渡金属催化的一些最重要的生化过程,它们也可以作为新型的仿生催化灵感。在后者的情况下,这些研究人员想确定金属中心和蛋白矩阵分别为酶系统的催化活性。他们分析了IPN的催化机制,三肽底物的转化势能面探索δ-(l—α- aminoadipoyl)—l半胱氨酰—D缬氨酸(ACV)对异青霉素N(IPN)。在高斯09 ONIOM设施使过渡态结构和反应是涉及这样大的蛋白质反应系统计算出的路径。
2、预测和解释光谱
光谱是研究分子结构和性质的基本工具。然而,观察到的光谱往往难以解释。电子结构的计算结果可以对这个过程至关重要。例如,预测的光谱可以被检查以确定峰分配中观察到的光谱以及峰的位置和强度的比较与实验数据。高斯09可以计算相应的光谱常数和优良的准确性相关的分子特性。本文结合实验观测和理论计算可以得到非常准确的感兴趣的化合物的结构、光谱数据。高斯09可以预测各种光谱包括红外和拉曼光谱,核磁共振谱、紫外-可见,振动的圆二色性(VCD)、拉曼光学活性(ROA)、电子圆二色谱(ECD),旋光色散(ORD),超精细谱(微波),康登,赫茨伯格,康登/赫茨伯格出纳员出纳分析。
3、模拟核磁共振
高斯09继续加强计划的核磁共振功能。自旋-自旋耦合常数产生定量的最困难的光谱数据。的计算精度是高度依赖于基础设置。而量子化学的标准基组是价电子的发达,一个更复杂的电子密度接近核的描述是预测费米接触需要(FC)长期(通常的自旋-自旋耦合常数的最大组成部分)。研究人员在高斯,公司已深入这个问题的探索和开发了改性基组适合建模这些量DFT框架内;其结果总结如下。当被请求时,高斯09将自动执行两步计算NMR自旋-自旋耦合,采用集总的计算和相应的修改的基础上设置FC术语标准的基础
4、研究手性
手性分子在许多研究中的重要性。高斯09可以研究手性与包括最新的两种光谱课的几种技术:VCD和ROA。例如,研究人员使用了VCD设施模型螺旋肽。由丙氨酸肽结构为主α螺旋的C末端被线圈状。VCD用同位素标记实验证明α-螺旋翼二十在ALA二十五(上图右)noncooperatively细线从温度升高,结束。
在高斯09研究这些系统的IR和VCD光谱。结果ALA二十五出现以下。计算成功地再现了实验观测,还能够量化的程度的“磨损”。他们还表明,信心可以放在报道的分子结构。
5、光学光谱预测
稳态光谱是研究平衡结构和不同的电子态的势能面最基本的工具之一。然而,解释这些实验数据往往是不简单。这种情况通常很大,计算有助于解释和分配各光谱特征的效益。几个高斯09能力的研究。例如,含时密度泛函方法生产高质量的激发态系统的描述(相当于DFT基态),和康登和赫茨伯格泰勒分析可用于计算从基态和激发态的电子跃迁频率的振幅分析。两者的结合可以有效地治疗转变的大的振子强度和禁戒跃迁。溶剂化效应可以被包括在这些模型。这些方法已被用来计算赫茨伯格特勒吸收和荧光QX自由基卟啉的光谱。他们的研究结果证明的权利。这些图比较高清晰度拟吸收和发射带,绘图计算和实验强度除以ω(吸收)或ω三(排放),并显示出良好的协议。
6、超精细光谱预测
高斯09计算最重要的张量,这有助于超精细谱。计算表明在该地区寻找过渡,使实验更有效。理论结果还可用于观察峰频谱分配,它可以是困难的或不可能完全确定从原始实验数据。计算张量也可以结合拟合操作观察。通过计算有助于解释观察到的结果应该和拟合非线性分子服从线性的研究。开壳层的物种特别丰富和复杂时,微波光谱研究。这种化合物在许多情况下是非常重要的,包括星际介质的化学。研究人员研究了1,1-difluoroprop-2-ynyl基、F二CC≡CH,部分氟化的变种的炔基。所观察到的微波光谱数据和各种超精细张量计算确定该化合物具有平面结构的组合,一个有点意外的结果。
7、热化学
准确的预测ΔG是理解许多化学反应至关重要。高斯09提供预测热化学量各种非常精确的能量的方法,包括完整的基组(CBS)的方法,该方法通过gaussian-4 gaussian-1家庭,和W1的方法。除了ΔG和ΔH,你可以预测地层,雾化能量,电离电位加热,电子亲合能和质子亲和力的范围广泛的化合物,在最高精度。
8、光化学和其他激发态过程
除了要考虑早期的光谱特征,高斯09还提供了研究激发态系统等几个特点。使用这些功能,你可以研究染料和其他的光吸收特性,杀虫剂和其他化合物的光解速率,对太阳能的利用材料的性能潜力,和许多其他同样重要的研究问题。高斯09研究这样的反应提供了许多方法,适用于不同大小的系统,包括随时间变化的DFT方法,该方法(EOM-CCSD在精度和耦合簇单打和双打的基态计算成本比较),和CASSCF方法提供了一种多分子系统的描述,也可以用来定位锥形交叉在激发态的基态势能面。
9、溶剂效应
可以考虑在优化结构和分子特性预测。例如,VCD研究丙氨酸肽链以页4-5是在溶液中进行。事实上,在这种情况下,气相的结果是非常不同的溶剂效应是研究这个系统的关键。高斯09的溶剂化能力是最先进的,他们提供大致相当的性能的气体研究许多计算类型相。
gaussian09w d.01软件适用范围
1、化学反应过程,如稳态及过渡态结构确定、反应热、反应能垒、反应机理及反应动力学等;(详情请点击)化学
2、各类型化合物稳态结构的确定,如中性分子、自由基、阴、阳离子等;(详情请点击)药学
3、各种谱图的验证及预测,如IR, Raman, NMR, UV/Vis, VCD, ROA, ECD, ORD, XPS, EPR, Franck-Condon及超精细光谱等;(详情请点击)环境
4、分子各种性质,如静电势、偶极矩、布居数、轨道特性、键级、电荷、极化率、电子亲和能、电离势、自旋密度、电子转移、手性等;(详情请点击)食品
5、热力学分析,如熵变、焓变、吉布斯自由能变、键能分析及原子化能等;(详情请点击)生命
6、分子间相互作用,如氢键及范德华作用;(详情请点击)药学
7、激发态,如激发态结构确定、激发能、跃迁偶极矩、荧光光谱、磷光光谱、势能面交叉研究等
gaussian 09 win 64位基本内容
一、基本算法
1、计算1 - &电子积分在任何合同的高斯函数
2、传统的、直接的、半直接和核心内的算法
3、线性化计算成本通过自动化的快速多极子方法(FMM)和稀疏矩阵技术
4、网络/集群和共享内存(SMP)并行性
5、哈里斯初始猜测(更准确,尤其是对金属)
6、初始猜测猜测或片段自洽场解决方案生成的片段
7、密度拟合和库仑引擎纯DFT计算,包括集自动生成合适的基础
8、O(N)的换取高频DFT和混合
9、1 d,2 d、3 d周期性边界条件(PBC)能量和梯度(高频和DFT)
10、选择修剪网格数值积分, 其中包括设计非常高的精度
二、几何优化和反应建模
1、几何优化的平衡结构,过渡结构和更高的鞍点,在多余的内部,内部(Z-matrix),笛卡尔,或混合内部和笛卡尔坐标
2、多余的内部协调算法为大型系统设计,半经验优化
3、新的内部协调处理增强优化可靠性
4、牛顿和同步Transit-Guided拟牛顿(QST2/3)定位方法过渡结构
5、IRCMax过渡结构的搜索
6、放松和unrelaxed势能面扫描
7、新的实现内在的反应路径跟踪(IRC), 适用于ONIOM QM:毫米与成千上万的原子
8、反应路径优化
9、BOMD分子动力学(所有分析梯度方法);ADMP分子动力学:高频,DFT,ONIOM(莫:毫米)
10、优化的锥形十字路口通过state-averaged CASSCF
三、振动分析
1、振动频率和正常模式, 包括显示输出限制指定原子/残留/模式(可选模式排序)
2、可重新开始的分析高频和DFT频率。
3、非谐频率分析包括 完全不和谐的红外intensitie,DCPT2 HDCPT2 resonance-free方法计算非谐频率和配分函数,和电子振动的计算包括儿童早期开发
4、莫:MM ONIOM频率包括电子嵌入
5、分析红外和 静态和动态拉曼强度(高频& DFT;MP2 IR)
6、Pre-resonance拉曼光谱(高频和DFT)
7、预计频率垂直于反应路径
8、核磁共振屏蔽张量& GIAO磁脆弱的感情(高频,DFT,MP2)和 增强自旋自旋耦合(高频,DFT)
9、专业基础集核磁共振自旋自旋耦合计算
10、振动圆二色性(VCD)旋转的优势(高频和DFT)
11、动态拉曼旋光性(ROA)强度
12、拉曼和ROA计算可以分开进行频率分析与更大的基础设置(如推荐的文献)
13、谐波振动转动耦合
14、增强不和谐的振动分析
15、不和谐的振动转动耦合通过微扰理论
16、阻碍了转子分析
四、分子性质
1、电子圆二色性(ECD)旋转的优势(高频和DFT)
2、静电势,电子密度,密度梯度,拉普拉斯算子,磁屏蔽和感应电流密度在一个自动生成的网格
3、通过十六极多极矩
4、人口分析 包括每个轨道分析指定的轨道
5、原子的指控:马利肯, Hirschfeld,CM5
6、Biorthogonalization分子轨道(生产相应的轨道)
7、静电potential-derived指控:可,CHelp CHelpG, 他
8、自然轨道分析和 自然过渡轨道
9、自然键轨道(NBO)分析,包括中科院工作的轨道
10、静电能量和费米接触条件
11、静态和频率相关分析极化率和推导(高频和 DFT); 数字2推导(高频;DFT w /分析第三引出。)
12、增强光学旋转和光学旋转色散(奥德)
13、超精细光谱部分:电子g张量,费米接触条件,各向异性费米接触条件,转动常数,偶极子超精细计算,四次离心畸变,电子自旋旋转张量,核电四极常数,核自▇旋旋转张量
14、Franck-Condon分析(光致电离)
15、ONIOM集成电和磁性
五、ONIOM计算
1、增强2和3层ONIOM能量,梯度层使用任何可用的方法和频率
2、可选的电子嵌入莫:MM能量,梯度和 频率
3、增强莫:MM ONIOM优化最小值 和过渡结构通过microiterations包括电子嵌入
4、支持IRC计算
5、ONIOM集成电和磁性
6、灵活的设施建设每层初始猜测,可选地包括从先前的计算结果(s)
7、计算原子电荷可用于毫米计算
8、一个外部程序可用于一个或多个图层,选择接收Gaussian-calculated数量
六、激发态
1、ZINDO、能量
2、CI-Singles能量、渐变和频率。
3、可重新开始的time-dep。高频和DFT能量 和梯度
4、SAC-CI能量和梯度
5、EOM-CCSD能量(可重新开始的);可选输入振幅计算一组较小的基础
6、Franck-Condon,Herzberg-Teller FCHT分析
7、时间使用塔姆丹科夫DFT计算近似
8、CI-Singles TD-DFT解决方案,包括可选的激发能规范范围
9、具体由各州完成励磁和de-excitations解决方案
七、自洽反应场溶解模型
1、新的实现极化连续模型(PCM)设施的能量,梯度和频率
2、溶剂的影响振动光谱、核磁共振和其他属性
3、溶剂效应对ADMP calc轨迹。
4、溶剂效应对ONIOM计算
5、增强激发态的溶剂效应
6、SMDδG模型溶解和分区系数
7、其他SCRF溶剂模型(高频& DFT):昂萨格能量,梯度和频率。等密度面PCM(I-PCM)能量和自洽等密度面PCM(SCI-PCM)能量和梯度
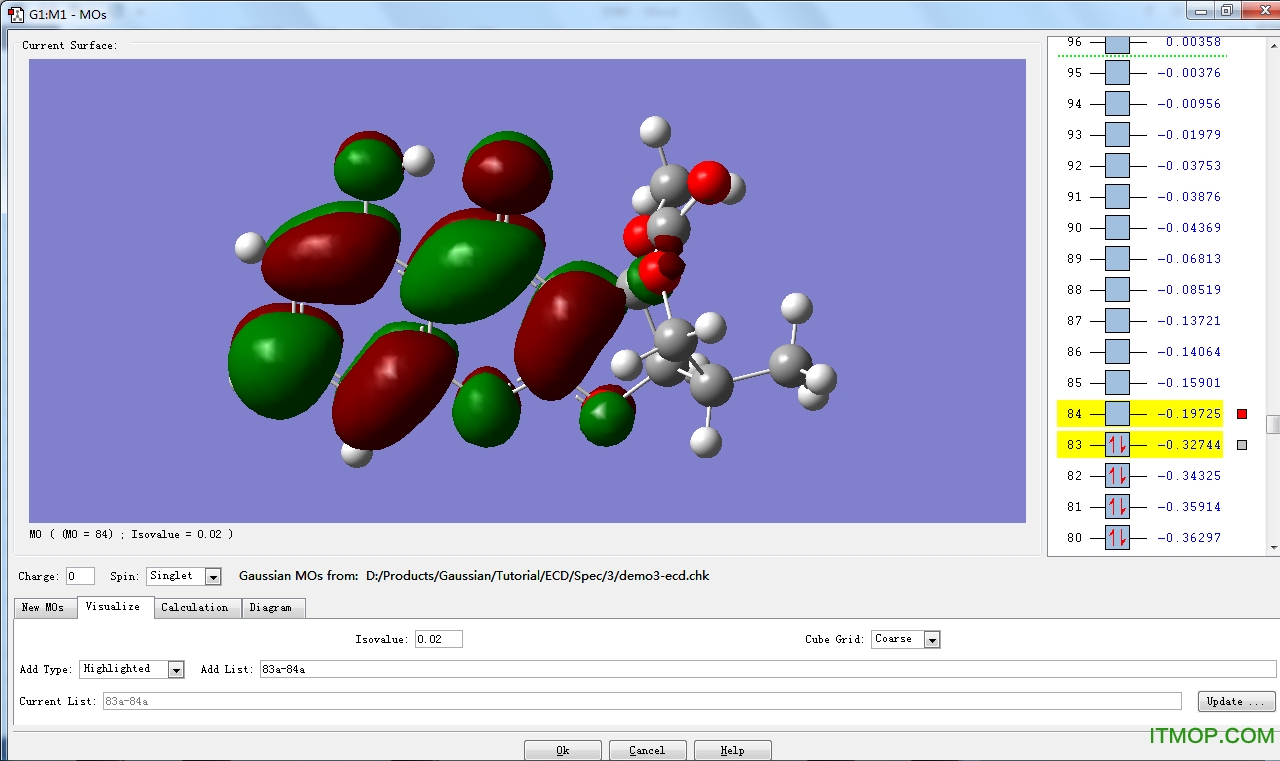
更多>> 软件截图
推荐应用

乐教乐学学生端电脑版 220.06 MB
下载/中文/9.0 v1.0.264 官方版
中国教育技术服务平台登录客户端 20.96 MB
下载/中文/10.0 v1.5 官方版
汉语大辞典完整破解版 50.20 MB
下载/中文/2.0 v6.70 最新免费版
金山词霸2005专业版 381.00 MB
下载/中文/7.0 超级完美版
奇迹英语背单词8.0破解版 219.00 MB
下载/中文/1.0 v8.0 中文免费版
堂堂网互动教学系统 17.75 MB
下载/中文/5.0 v2.3.18 官方版
北斗育才国学数字软件 109.09 MB
下载/中文/5.0 v16.21. 官方版
爱班云课堂 34.76 MB
下载/中文/10.0 v1.1 官方版
其他版本下载
精品推荐 化学绘图软件
 更多 (10个) >> 化学绘图软件 化学绘图软件是适用于化学、材料领域的学习软件,用户可以轻松绘制化学实验室设备、化学结构等.化学绘图软件可绘制化学分子结构、轨道、符号、箭头等图形,集成化学桌面办公软件,标准界面的菜单简明,易于操作,方便快捷!如chemdraw、InDraw、 Crystal Maker都是非常热
更多 (10个) >> 化学绘图软件 化学绘图软件是适用于化学、材料领域的学习软件,用户可以轻松绘制化学实验室设备、化学结构等.化学绘图软件可绘制化学分子结构、轨道、符号、箭头等图形,集成化学桌面办公软件,标准界面的菜单简明,易于操作,方便快捷!如chemdraw、InDraw、 Crystal Maker都是非常热
Crystal Maker(化学结构) 181.00 MB
/中文/10.0
indraw(化学结构绘图) 111.10 MB
/中文/5.0
KingDraw化学结构式编辑器 207.52 MB
/中文/8.0
仿真化学实验室 10.40 MB
/中文/10.0gaussian 09w 64位(高斯09程序包) 188.00 MB
/中文/0.0
化学金排12.0破解版(附注册码) 3.47 MB
/中文/0.0
ChemDraw Pro14.0(化学结构式画图软件) 93.09 MB
/中文/3.0
ChemBioOffice Ultra2015(化学结构绘制软件) 400.00 MB
/中文/10.0
相关文章
下载地址
gaussian 09w 64位(高斯09程序包) vd.01 破解版_(含中文手册+安装教程)
查看所有评论>> 网友评论
更多>> 猜你喜欢










下载周排行
下载总排行
- 教育学习精品热门应用

驾校一点通pc客户端 v7.0.0 官方桌面版
119.10 MB/中文/5.0

腾讯课堂pc客户端 v4.1.6.2 官方版
87.00 MB/中文/10.0

网易有道小图灵少儿编程 v1.3.1 官方版
80.77 MB/中文/5.0